
Alzheimer & Co in der Sackgasse – Epigenetik zeigt den Weg
Alzheimer & Co in der Sackgasse – Epigenetik zeigt den Weg
Knapp zehn Millionen Menschen sind allein in Europa an Demenz erkrankt. Dabei bestätigt sich auch, dass Demenz eine typische Erkrankung des weiblichen Geschlechts ist: auf 6,7 Millionen Frauen kommen 3,1 Millionen Männer mit Demenz.
In Zukunft werden immer mehr Menschen mit Demenz leben müssen: die Zahl der Erkrankten in Europa bis 2050 wird sich nahezu verdoppeln – die Gründe dafür sind das Bevölkerungswachstum und der demografische Wandel, der in vielen europäischen Ländern zu einem steigenden Anteil alter Menschen in der Bevölkerung führt. Demnach würden dann 14,3 Millionen Patienten in der EU und 18,8 Millionen Betroffene im weiter gefassten Euroraum leben.
Damit stehen den Gesundheitssystemen große Herausforderungen bevor.
Die sporadische Alzheimer Krankheit (LOAD) ist eine der häufigsten neurodegenerativen Erkrankungen weltweit. Trotz intensiver Forschungsbemühungen kann all diesen Patienten bisher nur unzureichend geholfen werden. Denn es gibt zwar Medikamente, die den fortschreitenden Hirnschwund etwas bremsen können. Doch heilen lässt sich diese Form der Demenz bislang nicht, darum setzen Ärzte auf Prävention und frühe Intervention.
Juni 2021 wurde in den USA ein neues Medikament gegen Alzheimer zugelassen – das erste neue Medikament seit dem Jahr 2002. Für viele Betroffene ist dies der lang ersehnte Hoffnungsschimmer im Kampf gegen die Krankheit des Vergessens.
Gleichzeitig zeigt es jedoch, wie weit wir nach wie vor von einer Heilung der Alzheimer-Krankheit entfernt sind. Denn auch Aduhelm von Biogen kann Alzheimer weder heilen noch den Krankheitsverlauf stoppen. Allenfalls ermöglicht es eine leichte Verzögerung der kognitiven Einbußen.
Das Erkrankungsrisiko kann gesenkt werden, heilen lässt sich Alzheimer bislang nicht
Da es noch kein Heilmittel für Alzheimer gibt, wird das Thema Prävention in der Alzheimer-Forschung immer wichtiger.
Studien zeigen, dass Menschen seltener an Alzheimer erkranken, die sich regelmäßig bewegen, sich geistig fit halten, gesellig sind, sich gesund ernähren und auf mögliche gesundheitliche Risikofaktoren achten und auch behandeln lassen, wie Bluthochdruck, Diabetes, Herzrhythmusstörungen und erhöhte Cholesterinwerte. Rauchen, Drogen- und übermäßiger Alkohol-Konsum und Übergewicht sind zu vermeiden.
Epigenetik zeigt den Weg
Warum fehlt es seit Jahrzehnten an Fortschritten in der kausalen Alzheimertherapie?
> Bislang verhindert die gezielte Suche nach einem einzelnen wirksamen „Heilmittel für Alzheimer-Kranke“ die Lösung des Problems. Es gibt keine Panazee, denn diese illusorische Universal-Lösung aller Probleme ist nur dem Wunschdenken der Pharmakonzerne und Value-Investoren geschuldet.
> Das Handikap der geförderten wissenschaftlichen Grundlagenforschung, da Studien bei neurodegenerativen Erkrankungen zeitlich zu kurz greifen.
> Die Ratlosigkeit über die ersten Ursachen und den frühen Verlauf der Alzheimer-Krankheit.
> Sich ergebende Folgen im Krankheitsverlauf werden als Ursachen der Alzheimer-Krankheit dargestellt.
Der Großteil der Forschung zum Verständnis und zur Behandlung von Alzheimer hat sich auf die „Amyloid-Hypothese“ konzentriert. Riesige Geldsummen wurden in Experimente mit Mäusen investiert, die genetisch verändert wurden, um Amyloid zu produzieren, und in die Entwicklung von Arzneimitteln, die Amyloidproteine blockieren oder zerstören oder manchmal Tau-Verwicklungen abbauen.
So ist das Mäusehirnmodell für die Erklärung der ersten Ursachen der Alzheimer-Demenz-Pathologie bei Monoaminooxidasen-Überexpression als Starter der mitochondrialen- und BBB-Dysfunktion völlig ungeeignet, da sich das menschliche Gehirn und das Gehirn dieser Nagetiergattung deutlich im Gehalt und der Verteilung der Monoaminooxidasen B und A unterscheiden und falsche Schlüsse zulässt.
Es ist klar geworden, dass dieser Ansatz nicht funktioniert. Alleine im Jahr 2018 gaben die US National Institutes of Health 1,9 Milliarden US-Dollar für Alzheimer-Forschung aus. Einer kürzlich durchgeführten Studie zufolge lag die Ausfallrate der Arzneimittelentwicklung bei Alzheimer jedoch bei 99 Prozent.
> Die „Prävention“ der Alzheimer-Krankheit erschöpft sich im epigenetischen Denken auf das Exposom der Alzheimer-Krankheit und derzeit auf die Hoffnung der Produktion von Histondeacetylase-Hemmstoffen, die für Pharmakonzerne dann ein für sie monopolisierbares Produkt am Markt sein könnte.
Der potente Weg der altersabhängigen Transmethylierung mit dem singulären Methylgruppen-Donor für die DNA, dem S-Adenosyl-L-Methionin (Ademetionin), der für die Stummschaltung der Monoaminooxidasen B und A bei Monoaminooxidasen- Überexpression sorgt und toxische Folgen an Mitochondrien (ATP), granulärem endoplasmatischem Retikulum (Proteinsynthese) und Lysosomen (Autophagie) verhindert, wird einfach negiert.
So bleiben die Endpunkte der Alzheimer-Krankheit, Neurodegeneration und Neuroinflammation, unbeeinflusst.
Drei Metabolite des Stoffwechsels schützen vor Alzheimer-Krankheit
- S-Adenosyl-L-Methionin (Ademetionin) aus dem Ein-Kohlenstoff-Zyklus,
- Spermidin aus der Aminopropylation des Ein-Kohlenstoff-Zyklus, wobei S-Adenosyl-L-Methionin für seine Biosynthese unentbehrlich ist und
- Melatonin, dessen Biosynthese ebenso von der Anwesenheit von S-Adenosyl-L-Methionin abhängig ist.
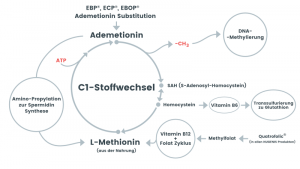
Abb. Der Ein-Kohlenstoff-Zyklus mit Transmethylierung, Transsulfurierung und Amino-Propylation
Die Biosynthese und die biologischen Wirkungen von Ademetionin, Spermidin und Melatonin sind zeitlich begrenzt und vom Altern abhängig.
Ademetionin, Spermidin und Melatonin haben gemeinsam, dass ihre Bioproduktion bereit im mittleren Erwachsenenalter deutlich abnimmt und die Substanzen substitutionspflichtig werden.
Die Folgen des Mangels an S-Adenosyl-L-Methionin (Ademetionin), Spermidin und Melatonin
-
der S-Adenosyl-L-Methionin (Ademetionin) – Mangel
Ein Ademetioninmangel führt zu Hypomethylierung (mangelhafte Versorgung mit Methyl-Gruppen) der Gen-Orte am Genom (Erbsubstanz) und zur Expression von Alzheimer-induzierenden Proteinen.
> Alzheimer-induzierende Proteine (Tau- und Amyloid-Pathologie):
- DSCAML1-Gen (Down syndrome cell adhesion molecule-like protein 1. Gene) exprimiert ein Protein im sich entwickelnden Nervensystem, wobei der höchste Expressionsgrad im fötalen Gehirn auftritt. Eine Überexpression des Proteins im sich entwickelnden fetalen Zentralnervensystem führt zum Down-Syndrom.
- BACE1 (Beta-Site-Amyloid-Vorläuferprotein-spaltendes Enzym 1), es ist für die Erzeugung aller monomeren Formen von Amyloid-β (Aβ) einschließlich Aβ 42 erforderlich. Die BACE1-Konzentrationen und -Aktivitätsraten sind in Alzheimer-Gehirnen und Körperflüssigkeiten erhöht, was die Hypothese stützt, dass BACE1 eine entscheidende Rolle in der Alzheimer-Pathophysiologie spielt.
- Presenilin-1 ist ein Presenilin-Protein, das beim Menschen vom PSEN1-Gen kodiert wird. Presenilin-1 ist eines der vier Kernproteine im Gamma-Sekretasekomplex, von dem angenommen wird, dass es eine wichtige Rolle bei der Erzeugung von Amyloid Beta aus Amyloid-Vorläuferprotein spielt.
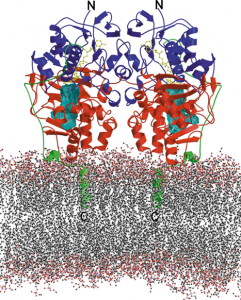
- MaoB und MaoA (MAOB-und MAOA-Gen) Monoaminoxidasen sind mitochondriale Enzyme, die Monoamine durch Desaminierung mit Hilfe von H₂O und O₂ zu den entsprechenden Aldehyden, Ammoniak und Wasserstoffperoxid abbauen,
- sind der Anlass für ROS und RNS. Die Belastung durch „Sauerstoffradikale“ und hochreaktive Stickstoffverbindungen leiten die Neurodegeneration ein,
- der mitochondrialen Dysfunktion,
- der TAU-Pathologie (AT8-ir Pathologie): Die Bildung von neurofibrillären Tangles unter oxidativem Zellstress beginnt vor der Pubertät oder im frühen jungen Erwachsenenalter in ausgewählten subkortikalen Kernen und nicht in der Großhirnrinde. Die Entwicklung der TAU-Pathologie geht der Entwicklung der Amyloid-β Pathologie um drei Jahrzehnte voraus.
- am rauen endoplasmatischen Retikulum stören MAOB und MAOA die Biosynthese der Proteine (Translation) und
- behindern den Autophagie-Prozess durch direkte Alteration der Lysosomen
- sie sind die Auslöser der Bluthirnschranken-Dysregulation und der darauf folgenden Neuroinflammation
- DKK1 (Dickkopf-related protein 1 ) Bei Alzheimer-Patienten wurde eine Herunterregulation dieses Wnt-Signalwegs als Folge hoher DKK1-Spiegel gezeigt. Aufgrund der durch DKK1 induzierten Hyperphosphorylierung kann Tau nicht mit neuronalen Mikrotubuli interagieren, wodurch der axonale Transport beeinträchtigt wird, was zu synaptischem Verlust und neuronaler Apoptose führt. Aufgrund seiner antagonistischen Wirkung auf den Wnt-Signalweg wird angenommen, dass DKK1 ein häufiger Marker für den neuronalen Tod bei neurodegenerativen Erkrankungen wie Alzheimer ist.
Homozystein ist ein Methylierungshemmer (DNMT’s) und stört den Osteogeneseprozess. Im Kohlenstoff-1-Zyklus bedarf es die Anwesenheit von Vitamin B6, Vitamin B12 und Folat um den Homozystein-Spiegel im Normbereich zu halten.
> Bluthirnschranken-Dysregulation und Neuroinflammation
Die Bluthirnschranke, brain–blood barrier (BBB) = neurovaskulären Einheit (NVU) ist wie eine Trennwand zwischen den Blutgefäßen und den Gehirnzellen (Neurone und Gliazellen). Sie ist wichtig, um die Bedingungen im Gehirn möglichst gleich zu halten.
Mikrovaskuläre Endothelzellen bilden Monoschichten, die ein integraler Teil der hochspezialisierten Bluthirnschranke sind. Mikrovaskuläre Endothelzellen sind durch Junctional-Proteine an einer Basalmembran befestigt und verschaltet. Zusammen mit mesenchymartigen Zellen, den Perizyten, glatten Muskelzellen, Astrozyten mit Endfeet-Fortsätzen und zirkulierenden Blutzellen bauen sie die sogenannte neurovaskulären Einheit (NVU).
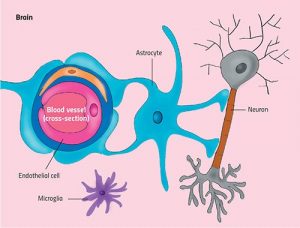
Abb. Gehirnendothelzellen bilden gemeinsam mit Neuronen und anderen spezialisierten Zellen (z.B. Ependymzelle, Neuron, Oligodendrozyt, Astrozyt, Mikroglia) ein interaktives Netzwerk, das als neurovaskuläre Einheit (NVU) bezeichnet wird. Quelle: Neuwelt, E. A., et al. Engaging neuroscience to advance translational research in brain barrier biology. Nature reviews. Neuroscience. 169-182 (2011).
Die neurovaskuläre Einheit (NVU ist eine dynamische, hoch spezifische und geregelte Schnittstelle, die den Übergang von Fluiden, Moleküle und Zellen zwischen zerebralen Blutgefäßen und dem ZNS steuert.
Eine Dysregulation der neurovaskulären Einheit kann zu einer Vielzahl von neurovaskulären, infektiösen, entzündlichen oder degenerativen Erkrankungen des ZNS, wie beispielsweise Schlaganfall, HIV-Enzephalopathie, multiple Sklerose, Morbus Alzheimer oder Morbus Parkinson, beitragen.
Astrozyten sind für die Bildung und Aufrechterhaltung der Bluthirnschranke unerlässlich, indem sie Faktoren bereitstellen, die zu einer adäquaten Verbindung zwischen den Zellen der Bluthirnschranke und der Bildung starker Tight Junctions führen.
Wichtig ist, dass die neurovaskuläre Einheit durch das Vorhandensein von Tight Junctions zwischen Hirnkapillarendothelzellen und der Expression spezifischer polarisierter Transportsysteme gekennzeichnet ist. Auf der anderen Seite stellen protoplasmatische Astrozyten durch ihre Endfüße die Verbindung zwischen den Inhaltsstoffen im Blutfluss des Endothels und den Neuronen her und sind so wichtige Regulatoren bei der Bildung und Aufrechterhaltung der neurovaskulären Einheit.
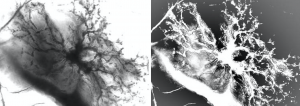
Abb. protoplasmatischer Astrozyt im orbitofrontalen Kortex des menschlichen Gehirns
Die erhöhte Monoaminooxidase B führt bei Alzheimer-Krankheit und zu den anderen neurodegenerativen Erkrankungen z.B: der Parkinson Krankheit zu zytotoxischen Dopaminabbauprodukten und in Folge zum Zelluntergang der protoplasmatischen Astrozyten. Damit fällt die Steuerungsfunktion des protoplasmatischen Astrozyten für die Hirnkapillarendothelzellen aus.
Die Stummschaltung von MAO B führt zu einer schützenden Wirkung vor dieser zellzerstörenden Bioaktivierung.
Eine gestörte Regulation der neurovaskulären Einheit kann zu Amyloid-Ablagerungen im Gehirn führen.
Bei Gehirntrauma werden gleichzeitig Astrozyten und Mikroglia aktiviert, was die Freisetzung von Zytokinen fördert, was wiederum die Endothelial Tight Junctions, die Pericyten beeinflusst und die Durchlässigkeit der neurovaskulären Einheit (NVU) für Fluids, Moleküle und Zellen erhöht.
Bei Alzheimer Krankheit finden sich zerebrale Amyloidangiopathien bzw. zerebrovaskuläre Amyloidosen und auch ausgedehnte Amyloid-Ablagerungen in der Groß- und Kleinhirnrinde (senile Plaques, Ablagerungen aus verklumpten Aβ-Peptiden, im Neuropil).
Es wird vermutet, dass der Eintritt von Proteinvorstufen über die neurovaskuläre Einheit aus dem Serum in das Gehirn bei der Alzheimer-Erkrankung zu Amyloid-Ablagerungen im Gehirn führt.
> Altern führt zu einem deutlichen Anstieg (Expression) von MAOB und seine Folgen
Der deutliche Anstieg (Expression)von MAOB im Alter um das 3 – 4-Fache geht parallel mit dem S-Adenosylmethionin (Ademethionin)-Mangel beim Älterwerden, der ab dem 35. Lebensjahr sich allmählich durch die Abnahme der S-Adenosylmethionin (Ademetionin)-Produktion in den Leberzellen entwickelt.
> Eine erhöhte MAOB-Expression findet sich:
- im Alter, hier ist die MAOB auf das 3 bis 4-fache erhöht,
- bei der Alzheimerkrankheit mit spätem Beginn (80% der Demenzen),
- bei toxischem Stress, Burnout und Depressionen,
- bei Rauchern,
- bei Alkoholabusus (regelmäßiger Konsum von Alkohol in einem gesundheitsschädlichen Ausmaß),
- bei Protonenpumpen-Hemmer (PPI)-Langzeiteinnahme und einer
- Langzeitcortisontherapie
Das Vorkommen der MAOB an der äußeren Mitochondrien-Membran, der Ort der MAOA an der inneren Mitochondrienmembran wie das Vorkommen der MAO B am Endoplasmatischen Retikulum und in den Lysosomen haben daher weitreichende Konsequenzen:
Im endoplasmatischen Retikulum kann oxidativer Stress ein Faktor sein, der die mPTP (ein Neurotoxin) – Übergangspore für die Mitochondrienpermeabilität auslöst und eine erhöhte MAO B Aktivität führt zu einer Stressantwort der Zellen mit Unterdrückung der Translation (der zweite Schritt der Proteinbiosynthese) durch fehlerhaft gefaltete Proteine.
- Wichtige Proteine sind der Komplex V der Atmungskette an der inneren Mitochondrienmembran, die ATPase. Ein insistierter ATP-Mangel hat für den gesamten Zell-Stoffwechsel schwerwiegende Folgen.
- Das Proteasom, ein Proteinkomplex, baut als Bestandteil der Proteinqualitätskontrolle bei Proteinfehlfaltung, Proteine zu Fragmenten ab. Proteasome sind mit der Autophagie für Recycling und Entsorgung von „Zellmüll“ , wie überflüssige (überschüssige) oder beschädigte zelluläre Makromoleküle (Proteine, Nukleinsäuren) verantwortlich. Sind weniger Proteasome vorhanden, überlasten das das Autophagie-System.
- Innerhalb von Neuronen, in Synapsen und entlang neuritischer Fortsätze bilden sich Autophagosomen und Endosomen, die eine ausgedehnte neuritische Dystrophie bei Alzheimer Krankheit zeigen können.
- Auch die Tubulation von Mitochondrien und Auto-Lysosomen ist entscheidend für die Aufrechterhaltung der Homöostase von Organellen.
- Ein Kinesin-Motor-Protein, Kif5B, die mitochondriale Außenmembran oder die autolysosomale Membran dekoriert und zieht, um tubuläre Strukturen zu bilden. Kif5B bewegt sich entlang von Mikrotubuli mit einer durchschnittlichen Geschwindigkeit von 49 bis 54 μm / min.
- Mikrotubuli sind ein Transportsystem in Dendriten und im Axon, zwischen Zellkörper, der den Zellkern enthält und der synaptischen Peripherie.
- Die neurodegenerativen Erkrankungen, wie beispielsweise, Morbus Alzheimer und Morbus Parkinson, lassen sich letztlich auf massive strukturelle Veränderungen an Synapsen zurückführen, die damit die Dysregulation/Dysfunktion bzw. den Ausfall dieser „krankhaften“ Strukturen erklären.
Weitere Funktion von S-Adenosylmethionin als Methylgruppen-Donor
Methylierung von Noradrenalin zu Adrenalin und Abbau der Katecholamine
Melatonin-Biosynthese und Serotonin-Abbau, siehe unten
Histamin-Abbau, Histamin ist ein exzitatorischer Transmitter im Gehirn
Biosynthese von Carnitin aus Lysin, Carnitin sorgt für die Verbrennung von langkettigen Fettsäuren (Mitochondrien)und Acetyl-L-Carnitin-Mangel ist verbunden mit Depression und Insulinresistenz. Acetyl-L-Carnitin -Gabe reduziert beide.
Biosynthese von Ubichinon, Bestandteil in Komplex I-III der Atmungskette
Methylierung von Cobalamin zu Methylcobalamin zur Verwendung als Co-Enzym an der Methionin-Synthase, wichtig für die L-Methionin-Bereitstellung im Ein-Kohlenstoff-Zyklus
Die Bedeutung von S-Adenosylmethionin (Ademetionin) als neuroprotektive Verbindung
S-Adenosylmethionin (Ademetionin) hat eine antioxidative Wirkung auf das Gehirngewebe. Die Wirkung wird sowohl als Hemmung der Lipidperoxidproduktion als auch als Verstärkung des endogenen Glutathion-Antioxidans-Systems angesehen.
S-Adenosylmethionin (Ademetionin) hat einen direkten Einfluss auf die Glutathion-S-Transferase-Aktivität. Da die Alzheimer-Krankheit mit einer verminderten Glutathion-S-Transferase-Aktivität, einer verminderten S-Adenosylmethionin (Ademetionin) und einer erhöhten S-Adenosylhomocystein (SAH) einhergeht, unterstreichen diese Ergebnisse die entscheidende Rolle von S-Adenosylmethionin (Ademetionin) bei der Prävention der Alzheimer-Krankheit.
Hyperhomocysteinämie korreliert mit einem erhöhten reduzierten / oxidierten Glutathionverhältnis im Gehirn, einer verringerten Glutathion-S-Transferase-Aktivität und einer erhöhten Lipidperoxidation.
S-Adenosylmethionin (Ademetionin) potenziert die Aktivität von Superoxiddismutase und Glutathion-S-Transferase und stellt die veränderte Lipidperoxidation von Glutathion und Erythrozyten im Gehirn wieder her.
Referenz:
De La Cruz JP1, Pavía J, González-Correa JA, Ortiz P, Sánchez de la Cuesta F. Effects of chronic administration of S-adenosyl-L-methionine on brain oxidative stress in rats. Naunyn Schmiedebergs Arch Pharmacol. 2000 Jan;361(1):47-52.
-
der Spermidin – Mangel
Während des Alterungsprozesses nehmen die intrazellulären Spiegel von Spermin und Spermidin sowie die enzymatische Aktivität von Ornithin Decarboxylase (ODC) tendenziell ab.
Die De-novo- Synthese von Spermidin und Spermin im Organismus braucht die Anwesenheit von S-Adenosylmethionin (Ademetionin).
Putrescin wird durch Spermidinsynthase durch Addition einer Propylamingruppe, die aus der Decarboxylierung von S-Adenosylmethionin stammt, in Spermidin umgewandelt. Anschließend wird Spermidin durch Sperminsynthase in Spermin umgewandelt, wobei eine zweite Propylamingruppe hinzugefügt wird (Amino-Propylation).
- Spermidinmangel fördert die Stickstoffmonoxid (NO)-Synthese und verursacht nitrosativen Stress. Nitrosativer Stress ist ein Zustand, der dann entsteht, wenn Zellen zu viel Stickstoffmonoxid (NO)-Gas bilden (Stickstoffmonoxid-Gas ist ein Botenstoff der Zellen).
- Spermidinmangel führt zu Störungen im Monoamin-Stoffwechsel: Hier werden Nervenbotenstoffe (Dopamin, Serotonin, Noradrenalin) beeinflusst, so dass Hirnfunktionsstörungen mit Einschränkung der Kognition und Gedächtnisleistungen, der Aktivität, des Antriebes bis hin zu psychiatrischen Symptomen (Depression) auftreten können.
- Im Zentralnervensystem beeinflussen Spermidin und Spermin unter anderem die Funktion von Glutamatrezeptoren vom NMDA-Typ und AMPA/Kainat-Typ, sowie einwärtsgleichrichtender Kaliumkanäle (Kir-Kanäle).
- Spermidin ist an der synaptischen Übertragung und der synaptischen Plastizität beteiligt, die dem Lernen und dem Gedächtnis zugrunde liegen.
- Den Effekt des Spermidin-Mangels kann man auch bei älteren Menschen beobachten, die unter Alzheimer-Krankheit leiden. Alzheimerpatienten haben im Durchschnitt 35 % niedrigere Polyamin-Werte als gleichaltrige Menschen mit gesunden Gehirnfunktionen.
- Auch auf das Schlafverhalten hat Spermidin einen Einfluss. Menschen die keine ausreichende Menge Spermidin zu sich nehmen, haben einen verlängerten circadianen Rhythmus. Diesen Effekt des Spermidin-Mangels kann man unter anderem auch bei älteren Menschen beobachten, die unter Alzheimer-Krankheit leiden. Alzheimerpatienten haben im Durchschnitt 35 % niedrigere Polyamin-Werte als gleichaltrige Menschen mit gesunden Gehirnfunktionen.
- Durch die Zufuhr von Ademetionin und Spermidin kann z.B. die Schlafarchitektur verbessert werden.
- Spermidin hat eine antidementielle Wirkung
- Bei der Alzheimer-Krankheit besteht eine Autophagie-Dysfunktion bei Spermidinmangel, die zu eine neuritischen Dystrophie durch Beeinträchtigung der Clearance von autophagischen Vakuolen führt.
- Spermidin schützt vor toxischem neuronalen Stress und schützt dabei die Gehirnzellen und den Gedächtnisapparat.
-
Toxischer Stress ein Risikofaktor der Alzheimer-Krankheit
Stress und eine überaktive Stress-Achse im Gehirn, HPA-Achse (hypothalamic–pituitary–adrenal- axis) genannt, spielen ebenfalls eine bedeutende Rolle bei der Alzheimerkrankheit.
Wo Stress aktiv ist, geht es immer auch um depressive Symptome und Schädigung des Lernzentrums im Gehirn, dem Hippocampus. Chronischer Stress gilt entsprechend auch als ein wesentlicher Risikofaktor für die Entwicklung der Alzheimerkrankheit, könnte aber auch eine Folge der Erkrankung sein (Gil-Bea und Kollegen, 2010 im Journal of Alzheimer’s Disease veröffentlicht). Stress schädigt aber auch die Mitochondrien, wie experimentell gezeigt werden konnte.
3) der Melatonin-Mangel
Schlafstörungen treten häufig bei der Alzheimerkrankheit auf. Forschungsergebnisse deuten an, dass gestörter Schlaf sogar zum Abbau der Denkleistung und der Entwicklung der Alzheimer-Krankheit beitragen, könnten.
Schlafstörungen stehen aber auch in engem Zusammenhang mit einer gestörten Funktion der Mitochondrien. Die Daten deuten auf einen Beitrag der Mitochondrienstörung zur Schlafstörung bei der Alzheimerkrankheit. Interessant ist in diesem Zusammenhang das umgangssprachlich als ‚Schlafhormon‘ bezeichnete Melatonin, das wesentlich zur Schlafregulation beiträgt, offenbar auch Veränderungen des Schlaf-Wach-Rhythmus im Verlauf der Alzheimerkrankheit bzw. bei noch leichten Einschränkungen der Denkleistung entgegenwirkt (Cardinali und Kollegen, 2010 im Fachjournal Current Neuropharmacology erschienen) und Schlafstörungen bessert.
Melatonin, ein aus der Zirbeldrüse synthetisiertes Neurohormon, das neuroprotektiv u.a. bei Alzheimer-Erkrankung ist. Hier spielt Melatonin eine entscheidende Rolle bei der Hemmung der circadianen Störung, indem es die Clock-Gene kontrolliert und die Aβ-Akkumulation und die Tau-Hyperphosphorylierung durch Regulierung des Signalwegs der Glykogensynthase-Kinase-3 (GSK3) und der Cyclin-abhängigen Kinase-5 (CDK5) abschwächt. Melatonin beeinflusst die Neurogenese, indem es die circadiane Störung, die Aβ-Bildung sowie die Tau-Hyperphosphorylierung abschwächt.
Melatonin zeigt seine neuroprotektiven Funktionen durch die Blockierung der Aβ-Produktion, Aβ-Oligomerisierung und -Fibrillation, Tau-Hyperphosphorylierung, synaptische Dysfunktion, oxidativen Stress und neuronalen Tod während der AD-Progression.
Gesund sein oder krank sein?
Unser Körper ist gesund, wenn es einen Ausgleich zwischen den gesundheitsfördernden und den krankheitsfördernden Einflüssen gibt. Trotz Störung kommt es vorerst zur Wiederherstellung der gesunden Ausgangssituation durch Selbstheilungskräfte.
Überwiegen aber die krankheitsfördernden Einflüsse, so wird eine Wiederherstellung der Unversehrtheit (Restitutio ad integrum) unmöglich.
Die Diagnose der Krankheit in Bezug auf ihre Erstursachen kommt immer zu spät. Zum Zeitpunkt der Diagnose der Krankheiten mit mitochondrialer Dysfunktion ist eine kausale Therapie des bereits vollentwickelten Organ/Systemdefektes mit einer Restitutio ad integrum (Heilung) unmöglich. Darum kann man auch nicht von Krankheitsvorsorge sprechen, wenn diese Krankheit bereits subdiagnostisch/subklinisch besteht.
Die Entwicklung von Biomarkern für Alzheimer, vor allem für die häufigste Form – LOAD (Late-onset Alzheimer’s disease) – wäre von entscheidender Bedeutung. Biomarker sind nicht nur bedeutsam für eine frühe Diagnose, sondern sie liefern auch Informationen über die Prozesse und Mechanismen, die der Krankheit zu Grunde liegen. Obwohl sich die Forschung seit einiger Zeit mit der Suche nach Biomarkern beschäftigt, sind noch keine akkuraten Biomarker für LOAD verfügbar.
Quellen:
Gender-related increase of tropomyosin-1 abundance in platelets of Alzheimer’s disease and mild cognitive impairment patients
Reumiller, CM., Schmidt, GJ., Dharami, I., Umlauf, E., Rappold, E., Zellner, M.
Journal of Proteomics (2017) (epub ahead of print)
Careful neuropsychological testing reveals a novel genetic marker, GSTO1*C, linked to the pre-stage of Alzheimer’s disease
Umlauf,E., Rappold, E., Schiller, B., Fuchs, P., Rainer, M., Wolf, B., Zellner, M.
Oncotarget 7 (26) (2016)
A platelet protein biochip rapidly detects an Alzheimer’s disease-specific phenotype
Veitinger, M., Oehler, R., Umlauf, E., Baumgartner, R., Schmidt, G., Gerner, Ch., Babeluk, R., Attems, J., Mitulović, G., Rappold, E., Lamont, J., Zellner, M.
Acta Neuropathologica 128(5) (2014)
Daher besteht immer ein Handlungsbedarf. Wir sind immer auf dem Weg zu unserem Gesundheitsziel. Die trügerische Illusion gesund zu sein und die alltägliche Sorglosigkeit begleiten uns ständig und von diesen getäuscht, werden wir oft erst nach Jahrzehnten von dieser Krankheit überrascht.
Vektorraum „gesund sein“
Die Werkzeuge der Epigenetik helfen diesen Balanceakt zwischen Physiologie und Pathologie zu Gunsten der Gesundheit (Physiologie) zu unterstützen.
Ganz allgemein könnte man unter Epigenetik die Wissenschaft von der Biochemie der Zelle und der Physiologie für den Organismus, als molekulare Informationsübertragung, die nicht durch Codierung der DNA übermittelt wird, verstehen.
-
DNA-Methylierung
Ademetionin (S-Adenosylmethionin) dient de facto als einziger Methylgruppendonor bei der Transmethylierung von DNA, RNA, Protein, Lipiden und Polysacchariden. Es wird geschätzt, dass die methylierte Schwefel (Sulfonium)-Einheit von Ademetionin gegenüber DNA und RNA 1000-fach reaktiver ist als andere Methylgruppendonatoren, wie die verschiedenen Formen von Folsäure oder Betain.
Alle Organismen synthetisieren S-Adenosylmethionin (Ademetionin, AdoMet) und ein großer Teil aller Gene sind Ademetionin-abhängige Methyltransferasen (Erhaltungs-MAT1A und De novo-MAT2A).
Die Ademetionin-abhängige Methylierung ist für viele biologische Prozesse von zentraler Bedeutung. Wachstumsfaktoren, Zytokine und Hormone, die Leber- Methyltransferasen und mRNA-Spiegel und die Enzymaktivität regulieren und bestimmen, dass S-Adenosylmethionin (Ademetionin) nicht nur als Zwischenprodukt im Methionin-Zyklus (Ein-Kohlenstoff-Zyklus) angesehen werden kann, sondern darüber hinaus auch als intrazellulärer Kontrollschalter in der Leberzelle, der wesentliche Leberfunktionen wie Regeneration, Zell-Differenzierung und die Empfindlichkeit dieses Organs gegenüber Verletzungen reguliert und zum Leberschutz dient.
Die DNA-Methylierung findet an der C5-Position von Cytosinresten am Genom statt. Die Hypermethylierung von CpG-Inseln definiert Regionen mit hoher CpG-Dichte, die in ungefähr 70% der Genpromotoren auftreten, die zu einer starken Gen-Stummschaltung führen.
non-Histonmethylierung
- Telemerase-Methylierung
- miRNA (2′-O-Methylierung. Da die chemischen Eigenschaften zwischen RNA und DNA liegen, wird angenommen, dass die 2′-O-Methylierung zu der reaktiven Gruppe von RNA-Molekülen auf der frühen Erde gehört, die zu DNA geführt hätte.
- tRNA- und rRNA- Methylierung (Translation)
- Metaboliten-Methylierung (Stoffwechselaktivität)
- Protein-Methylierung (posttranslationale Modifikation)
-
Histonmodifikation
Durch Acetylierung: Epigenetischer Marker ist die Acetylgruppe, die durch Histonacetylierung die Zellen vor oxidativem Stress und Schädigung schützen kann. Der Einfluss der Histondeacetylase (HDAC)-Hemmung auf die Altersmerkmale steht im Allgemeinen im Einklang mit positiven Veränderungen für die Gesundheit des Menschen.
Durch Methylierung: Die Histonmethylierung ist aus genomweiten Assoziationsstudien zur Depression des Menschen bekannt. Toxischer Stress bei sozialen Niederlagen vermindert die Histonmethyltransferasen G9a und ein G9a-ähnliches Protein, welche H3K9me2 im Nukleus accumbens katalysieren. Die Überexpression von G9a im Nukleus accumbens wirkt antidepressiv.
Phosphorylierung, Ubiquitinierung und ADP-Ribosylierung sind weitere epigenetische Marker an den Histonarmen (Histonschwänzen).
-
Chromatin-Remodellierung
Chromatin-Remodellierung variiert die Eigenschaften des Erbguts ohne die Abfolge der Nukleotide zu ändern. Zusätzlich zu Proteinen wirken auch epigenetische Markierungen an der DNA (hauptsächlich durch DNA-Methylierungen) auf die Chromatin-Remodellierung ein. Neben der aktiven Regulation bei der Genexpression verleiht dynamische Remodellierung dem Chromatin eine epigenetische Rollen bei verschiedenen biologischen Schlüsselprozessen, wie DNA-Replikation und DNA-Reparatur in Eizellen, Apoptose, Chromosomen-Segregation, -Entwicklung und -Pluripotenz.
-
Mikro-RNAs
Ein weiteres zentrales epigenetisches Schaltersystem basiert auf der Produktion von kleinen RNA-Molekülen (Mikro-RNAs), die sogenannten nicht kodierenden RNAs.
miRNAs sind evolutionär konserviert, was darauf hindeutet, dass sie eine wesentliche Rolle bei der Transkriptionsregulation spielen.
MicroRNAs sind kurze RNA-Moleküle, die im Gegensatz zu gewöhnlichen RNA-Strängen nicht in Proteine übersetzt werden. Ihre Funktion besteht darin, sich an andere RNA-Moleküle anzulagern und dadurch deren Übersetzung in Proteine zu verhindern oder sogar den Abbau der RNA-Moleküle zu bewirken.
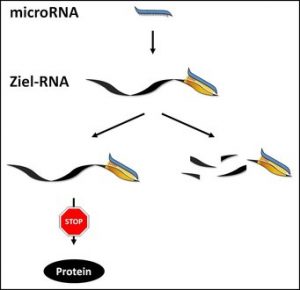
Abb. Eine microRNA lagert sich an die Ziel-RNA an und verhindert dadurch deren Übersetzung in ein Protein (links) oder löst deren Abbau aus (rechts). © MPI für Psychiatrie / Engel
miRNAs sind auch beteiligt bei der posttranskriptionellen Regulation von Nr3c1 (Glucocorticoid-Rezeptor). Interessanterweise ist in der Amygdala (Mandelkern)* die akute stressinduzierte Expression von mehreren miRNAs mit der Regulation der Angst mit Auswirkungen auf den Mineralocorticoidrezeptor und die corticotropin-releasing factor (CRF) -Expression verbunden.
* Amydala ist ein paariges Kerngebiet des Gehirns im medialen Teil des jeweiligen Temporallappens und ist Teil des limbischen Systems.
-
fasten-mimikrierende Substanzen als Alterungsprotektoren
Die wichtigsten Alterungsprotektoren sind vor allem die Ernährung, Fastenprozesse sowie fasten-mimikrierende Substanzen, welche die Signalkaskaden des Fastens einleiten, obwohl man nicht fastet.
Zu diesen gehören: Spermidin, Resveratrol, Rapamycin, Metformin und NAD-Metabolite, wobei das Pyridindinukleotid NAD+ ein Substrat aus Sirtuinen und Coenzym für Hydridtransferenzyme und andere NAD+ – abhängige ADP-Ribosetransferenzyme ist. Es existiert entweder in der Form von NAD+ oder NADH.
-
Neuroplastizität/Neurogenese
Das Phänomen der Neuroneogenese, also des Entstehens neuer Nervenzellen im erwachsenen Gehirn, spielt eine wichtige Rolle bei der Entstehung der Alzheimer Krankheit und eine gestörte Neuroneogenese ist ein wesentlicher aetiopathogenetischer Faktor, insbesondere für Demenzerkrankungen, Suchterkrankungen sowie schizophrenen und affektiven Psychosen.
Die adulte Neurogenese am Menschen wurde 1998 von Erikson und Mitarbeiter in Göteborg nachgewiesen.
In der subgranulären Zone des Gyrus dentatus, einem Teil der Hippocampus-Formation, als Region des ausdifferenzierten Gehirns, werden ständig Neurone neu gebildet. Der menschliche Hippocampus ist sein Leben lang in der Lage ist, Neurone zu erzeugen.
Wie wir heute wissen, kontrolliert eine große Palette von Faktoren die Neurogeneserate beziehungsweise die Lebensdauer von Neuronen im Gyrus dentatus.
Als neurochemische Stimulatoren beziehungsweise Inhibitoren der Neurogenese wurden Neurotransmitter, Wachstumsfaktoren und Hormone identifiziert, wobei hohe Konzentrationen an Cortisol die Neurogenese hemmt (Fuchs & Gould, 2000). Es wurde auch gezeigt, dass die Neurogenese durch körperlichen und sozialen Stress, Depressionen und eine Behandlung mit Antidepressiva gehemmt werden kann.
Der Hippocampus ist bei Lernen und Gedächtnis beteiligt. Er spielt jedoch auch eine wichtige Rolle bei der Regulierung der Stimmung.
Unter chronischem Stress und als Reaktion auf Corticosteronspiegel unterliegt der Gyrus dentatus einer Verringerung der Zellzahl, wohingegen körperliche Aktivität und ein angereichertes Umfeld das Volumen des Gyrus dentatus und die Neuronenzahl erhöhen.
-
Schutzwirkungen von Ademetionin und Spermidin im Allgemeinen:
durch Spermidin, das oxidativen Stress unterdrückt,
durch Spermidin, als ein wichtiger Wachstumsfaktor, der eine wichtige Rolle bei der Stabilität von Messenger-RNAs (mRNAs) und so auch Einfluss auf die Translation (Proteinsynthese), RNA-Bindung, RNA-Synthese, RNA-Synthese-Komplex und Regulation des Membranpotentials hat.
Spermidin-induzierte Autophagie
Autophagie ist die Aufrechterhaltung der Proteinhomöostasestase, also eines Gleichgewichts zwischen Synthese, Faltung und kontrolliertem Abbau von Proteinen. Sie ist für die Funktion jeder Zelle von entscheidender Bedeutung.
Die Entfernung fehlgefalteter, geschädigter, dysfunktionaler oder aggregierter Proteine kann grundsätzlich durch zwei Mechanismen erfolgen: das Ubiquitin-Proteasom-System und den Prozess der Autophagie.
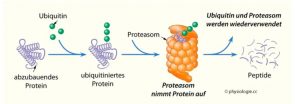
Abb. Proteasom als zellulären „Zerhexel-Maschine“ für Proteine
Bei der sogenannten Makroautophagie wird intrazelluläres Material (Proteinaggregate, Zellorganellen, Pathogene u.a.) von einer Membran umhüllt, die sich nachfolgend schrittweise zum Autophagosom schließt. Durch die sich anschließende Fusion mit einem Lysosom wird der Inhalt hydrolytisch abgebaut. Die Auswahl des abzubauenden Materials wird durch Rezeptormoleküle sichergestellt (selektive Makroautophagie), die einerseits mit dem Substrat, andererseits mit Proteinkomponenten der Autophagosomenmembran interagieren.
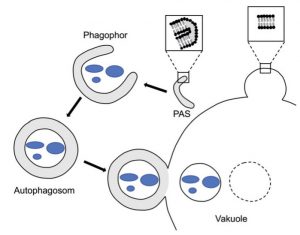
Abb. Autophagie
Vektorraum „krank sein“
Die neun Merkmale des Alterns:
Epigenetische Veränderungen, Telemer-Abrieb, Genomische Instabilität, Verlust der Proteostase, Deregulierte Nährstoffsignalisierung, Mitochondriale Dysfunktion, Zelluläre Seneszenz, Stammzellen Erschöpfung und veränderte interzelluläre Kommunikation.
- Das Altern als mitochondrialer Funktionsstörung
Die wissenschaftliche Evidenz, dass Altern mit Schädigung und Funktionsstörung der Mitochondrien einhergeht, ist überwältigend. Schon 1992 konnte in mehreren humanen postmitotischen Geweben (u. a. Gehirn und Muskel) gezeigt werden, dass es im Rahmen des Alterns zur Akkumulation von mtDNA-Mutationen und -Deletionen kommt, was zur Frage führte: „Growing old: the most common mitochondrial disease of all?“ (Harding 1992).
Diese mitochondriale DNA-Schädigung führt zu einer reduzierten Lebensspanne sowie zu vorzeitig einsetzendem Altern unterschiedlicher Organsysteme (zelluläre Seneszenz). Dieser Alterungs-Phänotyp ist verbunden mit einer kontinuierlichen Abnahme der Atmungskettenfunktion und einer verminderten ATP-Produktion.
- Epigenetik und Altern
Es zeigte sich, dass die Epigenetik ein Hauptregulator des Zellschicksals und der Zellfunktion ist und eindeutig die Gesundheitsbiologie (Physiologie), die Krankheitsbiologie (Pathologie) und die Langlebigkeit bestimmt.
Im Gegensatz zu DNA-Mutationen stellen epigenetische Veränderungen reversible Veränderungen dar und bieten das Potenzial für eine echte „verjüngende“ therapeutische Intervention (Anti Aging – Reverse Aging).
Epigenetische Signaturen ändern sich mit zunehmendem Alter auf natürliche Weise. Im Allgemeinen ist eine Abnahme der DNA-Methylierung im Genom (Hypomethylierung) zu verzeichnen, mit einer paradoxen Hypermethylierung bestimmter Genpromotoren im gesamten Genom.
Interessanterweise treten bei der Krebsentstehung dieselben DNA-Methylierungsmuster auf, die beim Altern beobachtet werden, wobei Tumorsuppressorgene durch Hypermethylierung ihres Promotors unterdrückt werden.
Veränderungen der Histonmodifikationen scheinen auch mit dem Alter aufzutreten. Es wurde gezeigt, dass in-vitro-Seneszenz mit einer Abnahme der repressiven Histonmark-Histon-3-Lysin-27-Trimethylierung sowie einer Zunahme der Methylierung von Histon-4-Lysin-20 einhergeht, was auf eine heterochromatische Region hinweist, wodurch die Gentranskription unterdrückt wird.
Diese Veränderungen der epigenetischen Muster können zu altersbedingten Krankheiten, Krebs oder einem verfrühten Alterungsprozess selbst beitragen.
-
Umwelteinflüsse und Verhalten
Umwelteinflüsse durch Armut, Unterernährung, toxischem Stress, mangelnde Bildung und gesundheitliche Ungleichheiten (Thayer und Kuzawa 2011; Brockie et al. 2013; Combs-Orme 2013; Owen et al. 2013) und körperliche Inaktivität, Rauchen, Alkoholkonsum oder Drogenkonsum können das Epigenom des Erwachsenen in Richtung Morbidität verschieben (Maccani und Knopik 2012).
Alterskrankheiten werden nicht durch einzelne genetische Defekte verursacht, sie resultieren vielmehr aus einer Kombination von Faktoren, einschließlich des quantitativen Beitrags mehrerer Gene (sogenannte quantitative Trait Loci, QTLs als Region eines quantitativen Merkmals auf einem Abschnitt eines Chromosoms für die Ausprägung eines quantitativen phänotypischen Merkmals) und Umweltfaktoren wie mangelnde Bewegung, toxischer Stress, inadäquate Ernährung, Rauchen, Drogen, Alkohol, mangelnde Bildung und gesundheitliche Ungleichheiten können das Epigenom des Erwachsenen in Richtung Morbidität verschieben.
Diese Faktoren führen zu einer Deregulierung verschiedener Signalwege, wie z. B. des Insulin-IGF1-Weges (Insulin-like Growth Factor 1), des Rapamycin (mTOR) -Pfades, des AMP-aktivierte Proteinkinase (AMPK) – Pfades und der Sirtuin-Pfade und können eine mitochondriale Dysfunktion induzieren.
-
Schutz der Gehirnzellen

EBP-Epigenetic Brain Protector enthält S-Adenosyl-L-Methionin zum Schutz der Gehirnzellen.
In der Entwicklung unserer Produkte legen wir zum Vorteil der AnwenderInnen größten Wert auf die Qualität der Inhaltsstoffe – daher verwenden wir beispielsweise Ademetionin in der höchst möglichen biologisch aktiven Form. Dasselbe gilt auch für die anderen Ingredienzen und deren Verarbeitung, die in ihrer Zusammensetzung die Gefahr der Hyperhomozysteinämie verhindern.
Für alle NUGENIS-Produkte liegen EU-konforme, österreichische Verkehrsfähigkeitsgutachten vor.

NUGENIS wurde 2015 auf der wichtigsten internationalen Fachmesse für „Ideen, Erfindungen, Neuheiten“, der iENA, für den EBP® – Epigenetic Brain Protector mit einer Goldmedaille für hervorragende Leistungen zum Schutz der Gehirnzellen ausgezeichnet.
NUGENIS Goldmedaille Iena 2015
Gefahr der Hyperhomozysteinämie, wenn der Ein-Kohlenstoff-Zyklus nicht begleitend durch die Substitution seiner wesentlichen Metabolite unterstützt wird
Homocystein gilt als neurotoxisches Oxidanz. An dem großen Patientenkollektiv der Framingham Study wurde prospektiv gezeigt, dass ein um eine Standard-Abweichung erhöhter Plasma-Homocystein-Spiegels mit einem um etwa 40% gesteigerten Risiko für das Auftreten der Alzheimer-Demenz assoziiert ist (Seshadri et al., 2002). Erhöhte Homocystein-Spiegel sind zusätzlich mit Hirnatrophie im Alter, Abnahme des hippocampalen Gewebevolumens und milder kognitiver Einschränkung sowie mit deren Uebergang in eine Alzheimer-Demenz assoziiert. Folsäure und Vitamin B12 sind Co-Faktoren bei der Remethylierung von Homocystein zu Methionin. Ein Folsäure-Mangel führt nach kurzer Zeit zu einer Erhöhung des Homocystein-Spiegels und ist mit einer Reduktion kognitiver Leistungen assoziiert.
Qualitätskriterien
Die Produkte der Angewandten Epigenetik von NUGENIS enthalten S-Adenosyl-L-Methionin (Ademetionin) in präventiver Dosis, um eine DNA-Hypomethylierung vorzubeugen und damit die sich ergebenden schwerwiegenden Schäden für den Organismus zu verhindern.
Die ausgewogene Zugabe von Vitamin B12, Vitamin B6 und Folsäure mindert die Gefahr einer Hyperhomocysteinämie.
Durch den hohen (S,S)–S-Adenosyl-L-Methionin (Ademetionin)-Anteil = aktive Form des S-Adenosyl-L-Methionin (Ademetionin) wird seine biologische Wirkung garantiert.
S-Adenosyl-L-Methionin (Ademetionin) wird bei der Einnahme vor dem Zugriff der Magensäure geschützt. Erstens durch eine magensaft-resistente Kapsel und zweitens als säurefestes gecoatetes S-Adenosyl-L-Methionin (Ademetionin)-Granulat.
Ihr Eduard Rappold
Alles was Sie über Ademetionin wissen sollten
Eduard Rappold
Eduard Rappold ist Autor, Unternehmer und als Arzt wissenschaftlicher Vermittler im Bereich Epigenetik und Präventionsmedizin. Im Zentrum seiner Arbeit steht die Frage, wie Umwelt, Verhalten und biografische Erfahrungen die Regulation unserer Gene beeinflussen – und welche Konsequenzen sich daraus für Gesundheit, Alterungsprozesse und chronische Erkrankungen ergeben. Sein Ansatz verbindet: aktuelle Erkenntnisse der Epigenetik neurobiologische Stressforschung mitochondriale und metabolische Regulation präventive und lebensstilbasierte Medizin Als Betreiber der Plattform epigenetik.at macht er komplexe wissenschaftliche Zusammenhänge für ein breites Publikum zugänglich. Dabei liegt der Fokus auf einer klaren, verständlichen Darstellung ohne Vereinfachung der Inhalte. Ein besonderer Schwerpunkt seiner Arbeit ist die Rolle von chronischem Stress als zentralem biologischen Faktor für Dysregulation, beschleunigtes Altern und Krankheitsentstehung. Eduard Rappold ist zudem Co-Autor einer wissenschaftlichen Studie zur Rolle von Antioxidantien und genetischen Faktoren bei neurodegenerativen Erkrankungen, insbesondere Alzheimer.